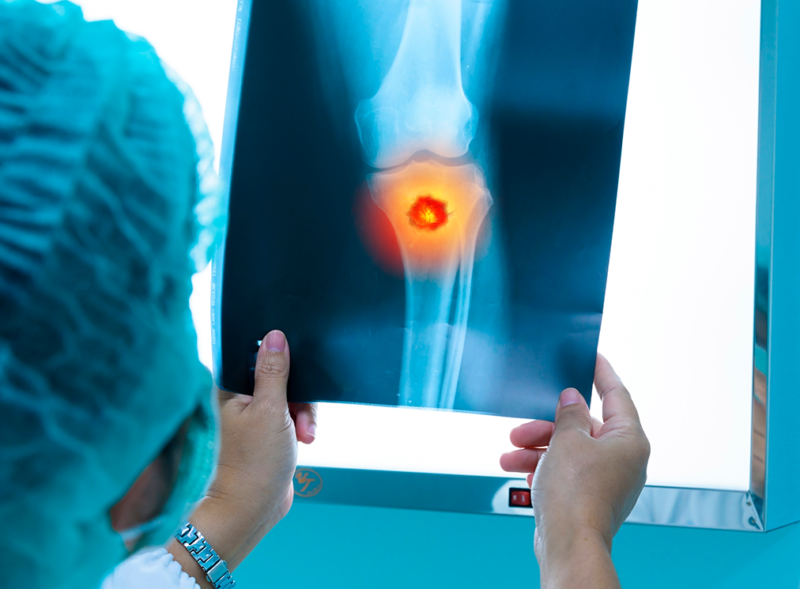
Según la ciencia, existe un tumor óseo maligno denominado sarcoma de Ewing que por lo general afecta a los adolescentes.
Se ha establecido, que se presenta con más frecuencia a partir de la segunda década de vida. Avances implementados en la actualidad, han permitido mejorar la tasa de supervivencia, pero, el porcentaje de recurrencia sigue siendo elevado.
De este tema, que preocupa a muchas familias voy a hablar a solicitud de Alina, una seguidora de Michigan. Ella es una dulce abuelita con un nieto de diez años, con quien ha compartido desde que era un bebé.
Ella me contactó muy preocupada porque desde mediados de enero, su nieto cuando se iba a la cama, manifestaba algunos dolores a nivel de los muslos y la pantorrilla.
Alina me comentó, que se sintió muy angustiada por ello y quiso llevar a su nieto al médico. Sin embargo, su hija le tranquilizó y le pidió que evitara las angustias. Ella me comentó, que su nerviosismo se debió a un caso de sarcoma de Ewing que ocurrió en su familia.
Esto a ella le produjo mucho nerviosismo desde niña y esto hizo que su nieto pudiera estar afectado.
A fin de tranquilizarla, le mencioné que de acuerdo a la ciencia esta enfermedad no era heredable. La prevalencia de esta enfermedad no es tan alta, pero se presenta en una población muy joven. Por eso consideré importante compartir este articulo con vosotros.
Generalidades acerca del sarcoma de Ewing
Según los expertos, el sarcoma de Ewing (SE) es un tumor agresivo que se presenta en adolescentes y adultos jóvenes. Estadísticamente, representa entre el 10% y el 15% de todos los sarcomas óseos. Esta enfermedad fue descrita por el médico estadounidense James Ewing en 1921, como un “endotelioma difuso de hueso”

Esta enfermedad está representada por el sarcoma de Ewing óseo “clásico” y el sarcoma de Ewing extraesquelético. Pero incluye además, el tumor maligno de células pequeñas de la pared torácica (o tumor de Askin) y los tumores neuroectodérmicos primitivos de partes blandas.
Según las investigaciones, los tumores de la familia del sarcoma de Ewing, presentan translocaciones cromosómicas no aleatorias. Es decir, una anomalía cromosómica estructural.
Los sitios del cuerpo más afectados por esta enfermedad, son el esqueleto axial, la pelvis, y el fémur. No obstante, puede presentarse en casi todos los huesos o tejidos blandos. Un síntoma común entre quienes padecen esta enfermedad, es el dolor y la inflamación de la zona afectada.
En las últimas cuatro décadas se han logrado importantes avances que han elevado los niveles de supervivencia. Así, se logró mejorar la tasa de supervivencia a 5 años inferior al 20% a más del 70%. Dentro de dichos avances destacan, la quimioterapia adyuvante multiagente y la terapia local. Sin embargo, su tasa de recurrencia es elevada.
Esta enfermedad se expresa localizadamente, sin embargo, la enfermedad metastásica subclínica ocurre de forma generalizada. Se ha determinado que, las tasas de supervivencia son menores del 30% en quienes presentan metástasis pulmonares aisladas.
Incluso, quienes presentan afectación de la médula ósea y de los huesos, presentan tasas de supervivencia menores al 20%. Por ello, es importante desarrollar nuevas terapias para lograr disminuir la metástasis (diseminación de las células cancerosas a otras zonas del cuerpo).
Ahondando acerca del sarcoma de Ewing
Hasta ahora, la ciencia no ha encontrado relación directa entre el SE y posibles factores de riesgo ambientales. Tampoco, se ha vislumbrado ninguna asociación entre esta patología y antecedentes de cáncer familiares, exposición a terapias farmacológicas o radiación.
El SE, se considera actualmente la segunda neoplasia ósea primaria con mayor frecuencia entre adolescentes y adultos jóvenes. La mediana de esta patología, es de 15 años de edad ello representa menos del 5% de todos los sarcomas de partes blandas.
De acuerdo a las estadísticas, en los Estados Unidos ocurren más de 200 casos anualmente. De hecho existen registros, que muestran que la incidencia del SE en el periodo de 1973 al 2004, fue de 2,93 por cada millón de habitantes.
Según los especialistas, el pico de incidencia oscila entre los 10 y 15 años de edad. Sin embargo, un 30% de los casos ocurre en niños menores de 10 años y otro 30% en adultos mayores de 20 años. Se ha observado además, que esta enfermedad se presenta mayoritariamente en el sexo masculino. Su proporción ha sido establecida en de 3 a 1 entre hombres y mujeres.
Síntomas y signos más frecuentes del sarcoma de Ewing
Las personas que padecen SE, presentan comúnmente durante semanas o meses, síntomas locales que incluyen, rigidez, dolor e inflamación. Incluso se presentan bultos palpables y más de la mitad de quienes padecen esta patología, sufren de dolores intermitentes que se incrementan durante la noche.

Los expertos sugieren, que ante la presencia de estos síntomas es importante acudir inicialmente al médico de cabecera. Una vez, que el haga la valoración se decidirá si amerita un examen por un especialista.
Esta enfermedad puede ubicarse en diversas zonas y mostrar presentaciones diferentes. Se ha observado, que se consigue con frecuencia en la diáfisis (porción central) de los huesos largos.
De hecho, las lesiones óseas o metastásicas en el interior de este hueso se observan como fracturas patológicas. A su vez, cuando el SE, se presenta a nivel de la pelvis, ocasiona dolor de espalda.
Quienes padecen SE presentan también ciertos síntomas sistémicos como fiebre (en un 25% de los casos) y pérdida de peso. Por lo general, cuando esto se presenta es indicativo de que existe enfermedad metastásica. Se ha establecido, que un 20% de los pacientes presentan enfermedad metastásica en el momento del diagnóstico.
Incluso, más del 20% de ellos muestran afectación a nivel de los pulmones o de la pleura. En estos casos, la persona afectada suele presenta ruidos respiratorios asimétricos.
En los casos que presentan metástasis en la médula ósea, puede observarse petequia (sangrado por ruptura capilar en la piel). Puede también ocurrir la presencia de púrpura por trombocitopenia, ocasionado por la disminución abrupta de las plaquetas sanguíneas.
Las personas con SE pueden también presentar diagnósticos semejantes a una fractura de huesos, ocasionada por un traumatismo. Incluso, puede ocurrir porque el tumor “se come” el tejido óseo. De hecho, en condiciones normales el hueso no hubiera llegado a romperse.
Pruebas más comunes realizadas para detectar el sarcoma de Ewing
Radiografía
Tan pronto el médico elabora la historia clínica (partiendo de las preguntas), y realiza el examen físico, solicita una radiografía. Este procedimiento permite valorar más detalladamente el área afectada. De hecho, se podrá apreciar si existe lesión osteolítica (aquella que causa la destrucción del hueso).
Dicha afección puede ocasionar daños en la zona (cortical) superficial del hueso o en los tejidos blandos. La destrucción del hueso, aparece con una apariencia laminada típica, semejando capas de cebolla.
Cabe destacar, que en las radiografías el SE de hueso luce como una lesión de crecimiento destructivo. Ello se ubica principalmente en la parte central del hueso, y técnicamente se denomina diáfisis.
Asimismo, otra característica visible empleando radiografía, es el aspecto “en rayos de sol”. Este efecto es producido por la neoformación de hueso a lo largo de los vasos del periostio. El periostio, es una membrana que contiene vasos sanguíneos y nervios, responsables de nutrir y otorgar sensibilidad al hueso.
Tomografía y escáner
Muchos especialistas cuando tienen la sospecha de la presencia de SE realizan adicionalmente ciertas pruebas de estadificación. Las mismas permiten determinar si el tumor se ha diseminado.
Por lo general, dichas pruebas son una tomografía computarizada (TC) de los pulmones y un escáner óseo. Mediante los resultados de estas pruebas, los médicos poseen mayor cantidad de información, logrando planificar adecuadamente y valorar el pronóstico.
Vale destacar, que en algunos centros médicos, en la actualidad se está empleando una nueva modalidad, denominada tomografía de emisión de positrones (PET). Sin embargo, el papel del PET en la evaluación y tratamiento de SE, no se ha definido completamente.
Según estudios realizados por la Universidad de Washington, se demostró que la respuesta al tratamiento valorada mediante el PET, puede predecir la supervivencia libre de progresión tumoral.
No obstante, estudios europeos han demostrado que para la evaluación del SE, resulta más efectivo emplear de forma asociada el PET y el TC. Según estos estudios, se obtiene una mejor precisión que empleando solo el PET aisladamente.
Biopsia
Una vez que todas las pruebas anteriores han sido realizadas, es imprescindible obtener una muestra del tejido del tumor. Esto se realiza, mediante una biopsia y permite confirmar que el tumor se corresponde con el SE.
De acuerdo a los expertos, existen dos posibles tipos de biopsia, la incisional y la excisional. Las biopsias incisionales, permiten obtener una pequeña muestra de tejido y pueden realizarse mediante agujas (biopsia cerrada) o mediante biopsias abiertas.
A su vez, las biopsias excisionales, se emplea cuando la masa es pequeña (es decir, menos de 5 cm) y no se encuentra cercana a estructuras críticas (paquetes neurovasculares).
Según los especialistas, es importante escoger adecuadamente el tipo de biopsia, una vez evaluado el tamaño y localización del tumor. Además de ello, debe considerarse la edad el paciente.
Incluso, debe escogerse adecuadamente el área donde se realizará la biopsia, incluyendo su relación, respecto a la ubicación del tumor. Se destaca además, la importancia de considerar las estructuras anatómicas del paciente.
Se ha determinado, que las lesiones pequeñas y superficiales pueden ser sometidas a biopsia excisional. Sin embargo, generalmente, si se sospecha la presencia de un tumor óseo maligno no se emplea este tipo de biopsia.
Ello obedece al hecho de que es frecuente que el tumor sea grande al momento del diagnóstico. Esto, hace necesario la presencia de terapia adyuvante (aplicación de quimioterapia) antes de extirpar el tumor.
Cabe destacar, que si el tumor, es probablemente benigno, considerando la historia clínica, examen físico y estudios de imagen, pero aún persisten las dudas, se practica una biopsia intraoperatoria.
Esta prueba permite realizar la valoración inicial rápida de una muestra congelada, mientras el paciente aún está en el quirófano.
Tratamiento del sarcoma de Ewing
Actualmente esta patología debe manejarse con la participación de facultativos de diversas especialidades médicas con experiencia en SE. Ello incluye oncología médica y pediátrica, traumatología oncológica, radiología musculoesquelética, oncología radioterápica y patología osteoarticular y de partes blandas.
Es crucial, que un paciente diagnosticado de SE sea tratado en un centro con mucha experticia en esta enfermedad.
Vale destacar, que los cirujanos generales, neurocirujanos, cirujanos vasculares y plásticos pueden ofrecer en ciertos casos un apoyo sustancial. Actualmente, el tratamiento de todos los sarcomas de Ewing (de huesos o de partes blandas), es el mismo. Existe una primera línea de tratamiento estandarizada, basada en diversos estudios. La misma consiste en:
- 14 a 17 ciclos de quimioterapia, alternados con dos regímenes de medicamentos
- Resección quirúrgica: mediante este procedimiento, cuando es posible hacerlo, se practica una cirugía conservadora de la extremidad afectada con reconstrucción mediante una prótesis. En caso de que exista una afectación del hueso, se realiza una implantación de hueso proveniente de un donante.
Según los especialistas, cuando la resección quirúrgica completa no es posible, podría necesitarse aplicar tratamiento radioterápico diariamente durante seis semanas.
El SE, es una neoplasia agresiva. Por ello, siendo conservadores, requeriría ser tratado durante nueve meses a un año. Si el tumor no responde al “tratamiento de primera línea” y aún se mantiene, pueden emplearse otros fármacos ya existentes. En el caso de que esto tampoco funcionase, la persona afectada podría ser llamado a participar en un ensayo clínico.
Últimamente, se ha mejorado significativamente la supervivencia, gracias a los avances en la quimioterapia. Luego de la cirugía, se aplica quimioterapia adicional, la cual se ajustará en función de la respuesta del tumor. En caso de que el tumor haya sido sensible a este tratamiento, en algunos casos se prevé un mejor pronóstico.
Concluyendo
De acuerdo a lo mostrado, el sarcoma de Ewing (SE) es un tumor agresivo que se presenta en adolescentes y adultos jóvenes.
Según los cálculos, este tumor representa entre el 10% y el 15% de todos los sarcomas óseos. Los estudios realizados hasta ahora, muestran que para acercarnos al pronóstico que puede tener esta patología, deben considerarse múltiples factores.
Entre ellos destacan, la edad del paciente, tamaño y localización del tumor, además de la respuesta a los tratamientos empleados. Vale destacar, que todos los aspectos del tratamiento han mejorado sustancialmente durante los últimos 30 años.
De hecho, ahora es tratada de forma multidisciplinar (quimioterapia, radioterapia y cirugía). Sin embargo, este continúa siendo una terapia altamente intensiva. En términos promedio, el tratamiento dura aproximadamente un año.
Durante este tiempo, el paciente y su familia, “invierten” parte de sus vidas, a fin de que los que quedan transcurran de forma esperanzadora.
Para Alina la información recibida, la cual comparto en el post, fue de gran apoyo. Su hija estuvo en el pediatra y este les informó que, los dolores de su nieto, eran debido al crecimiento.
Esto terminó de tranquilizarla, de hecho se mostró muy agradecida por mostrarle como la ciencia ha evolucionado tratando esta enfermedad.
“El sarcoma de Ewing es el segundo tumor óseo más frecuente de la infancia y la adolescencia, que también puede aparecer en tejidos blandos. El sarcoma de Ewing es un cáncer muy agresivo, con una supervivencia del 70-80% para los pacientes con riesgo estándar y enfermedad localizada y del alrededor del 30% para los que presentan enfermedad metastásica.”
Dr. Thomas G P Grünewald
Universidad de Múnich, Alemania
Si te ha gustado este artículo y tienes un interés sincero en aprender cómo puedes vivir más sano, me gustaría regalarte una copia de mi último libro #Yo Puedo con la Dra. Cocó.
Sí la página te da un mensaje de error es porque no has entrado la dirección bien. Vuélvelo a intentar, asegurando no haber dejado ningún espacio antes, después o entre las letras de tu dirección.
https://www.ncbi.nlm.nih.gov/books/NBK559183/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9842801/
https://pubmed.ncbi.nlm.nih.gov/17272319/
https://pubmed.ncbi.nlm.nih.gov/29977059/
https://pubmed.ncbi.nlm.nih.gov/31802230/
https://www.cancer.net/cancer-types/ewing-sarcoma-childhood-and-adolescence/statistics
https://medlineplus.gov/spanish/ency/article/001302.htm
http://sarcomahelp.org/ewings-sarcoma.html
https://www.mayoclinic.org/diseases-conditions/ewing-sarcoma/symptoms-causes/syc-20351071
https://www.cancer.gov/types/bone/patient/ewing-treatment-pdq